Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease) توسع الشعيرات النزفي الخلقي
INSTRUCTION
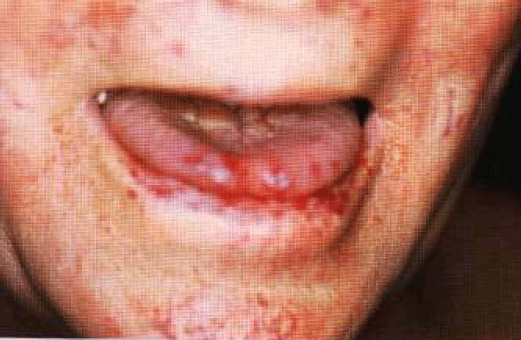
Examine the patient's face and obtain a relevant history.Perform a general examination.Look at this patient's face.
SALIENT FEATURES
History
Does it run in the family (autosomal dominant)?
Is there a history of gastrointestinal bleeding?
Is there a history of epistaxis?
Is there a history of repeated blood transfusions?
Is there a history of dyspnoea, fatigue, cyanosis or polycythaemia?
(pulmonaryarteriovenous malformations).· Is there a history of headaches, subarachnoid haemorrhage?
(cerebralarterio-venous malformations).Examination· Punctiform lesions and dilated small vessels present on the face, in particulararound the mouth · The patient may be pale (due to iron deficiency anaemia).
Proceed as follows
Look into the patient's mouth and inspect the tongue and palate for telangiectasia. ·Examine the nail beds, arms, trunk for telangiectasia.· Examine the chest for bruits (pulmonary arteriovenous malformations with apredilection for lower lobes).· Look for signs of cardiac failure caused by left-to-right shunting and hepaticbruits (both due to hepatic arteriovenous malformations) (N Engl J Med 2000; 343:938-52).
DIAGNOSIS
This patient has multiple telangiectases around the mouth and on the tongue and lips(lesion), probably hereditary in nature (aetiology). The patient is severely anaemic,probably as a result of upper gastrointestinal bleeding, and is currently receiving a bloodtransfusion (functional status).
QUESTIONS
What do you understand by the term 'telangiectasia'?
Telangiectasia is a cluster of dilated capillaries and venules. In this disordertelangiectases consist of focal dilatations of postcapillary venules.Mention a few conditions in which telangiectases are seen.Of the face:· Those who work outdoors in a temperate or cold climate (e.g. farmers).· In mitral stenosis.· Myxoedema.· Transitory phenomenon during pregnancy.Of other sites:· Secondary to irradiation.· Scleroderma (CREST syndrome).· Dermatomyositis.· SLE.· Acne rosacea.· Lupus pernio.· Polycythaemia.· Necrobiosis lipoidica diabeticorum.
ADVANCED-LEVEL QUESTIONS
What do you know about the genetics of hereditary telangiectasia?
Mutations in two genes, endoglin-beta (at chromosome 9q3; Nat Genet 1994: 6: 205-9)and activin receptor-like kinase 1 ALK-I (Nat Genet 1996; 13: 189-95), are known tocause HHT Each encodes a protein expressed on vascular endothelial cells andinvolved in signalling by members of the transforming growth factor (TGF)-[3superfamily. It is interesting that heterozygote endoglin-knockout mice develop aphenotype (J C/in hn'est 1999; 104: 1343-51) similar to that of humans who areheterozygous for a null mutation in the endoglin-[3 gene: the hereditary haemorrhagictelangiectasia. These heterozygous mice develop nose bleeds and cutaneoustel-angiectasia, and curiously the ears are more commonly affected than in humanbeings.
What do you know about the pathology of the condition?
In the skin· The small telangiectases are focal dilatations of postcapillary venules withprominent stress fibres in the pericytes along the luminal border.· In fully developed telangiectasia, there is marked dilatation of the venules, whichare also convoluted. They extend along the entire dermis, with excessive layers of smooth muscle devoid of elastic fibres. These often are directly connected to dilatedarterioles.· Mononuclear cells, predominantly lymphocytes, accumulate in the perivascularspace.In the lungs, liver and brainArteriovenous malformations lack capillaries and consist of direct connections betweenarteries and veins.
What are the clinical criteria for diagnosing hereditary haemorrhagic telangiectasia(HHT)?
Shovlin criteria· Recurrent epistaxis.· Telangiectasia at a site tither than in the nasal mucosa.· Evidence of autosomal dominant inheritance.· Visceral involvement.The diagnosis of HHT is definite if three criteria are present. A diagnosis of HHT cannot be established in patients with only two criteria, but should be recorded as possible orsuspected to maintain a high index of clinical suspicion. If fewer than two criteria arepresent, HHT is unlikely, although children of affected individuals should be consideredat risk in view of age-related penetration in this disorder (Am J Med Genet 2000: 91:66-7).
What are the complications of hereditary telangiectasia?
Epistaxis (usually begins by the age of 10 years and age 21 in most: it bec(linesmore severe in later decades in about two thirds of affected patients: N Engl J Med1995; 333: 918-24).· Gastrointestinal haemorrhage (usually does not manifest until the fifth or sixthdecade). Arteriovenous malformations, angiodysplasias and telangiectases are presentin the stomach, duodenum, small bowel, colon and liver.· Iron deficiency anaemia.· Haemoptysis, cyanosis, clubbing, cerebral abscess and embolic stroke due topulmonary arteriovenous malformations.· Headache and subarachnoid haemorrhage.· High-output cardiac failure is almost always associated with shunts from thehepatic artery to the hepatic veins (N Engl d Med 2000; 343:931-6).
How would you manage such patients?
Anaemia: ferrous sulphate, multiple blood transfusions.· Epistaxis: oestrogen, cauterization, septal dermatoplasty, laser ablation andtrans-catheter embolotherapy of arteries leading to the nasal mucosa.· Cutaneous telangiectasia: cosmetic therapy with topical agents, laser ablation.· Pulmonary arteriovenous malformations: embolotherapy, surgical resection orligation of arterial supply.· Gastrointestinal telangiectasia: blood transfusions, photocoagulation,oestrogen-progestogen therapy.· Brain and spinal cord arteriovenous malformations: embolotherapy,neurosurgery, stereotactic surgery.· Active bleeding: epsilon aminocaproic acid (N Engl J Med 1994; 330: 1789; NEngl J Med 1994; 330: 1822).r T his condition was described by H.J.L.M. Rendu, a French physician, in 1896,by Sir William Osier in 1901, and by F. Parkes Weber, a London physician, in 1936.